Saeij Lab
Host-Parasite Interactions and Toxoplasma gondii Pathogenesis at UC Davis
Welcome to the Saeij Lab at UC Davis, a leading Toxoplasma research lab
focused on the molecular and cellular basis of host–parasite interactions during
Toxoplasma gondii infection. Our research explores how this intracellular parasite causes Toxoplasmosis, how it
disseminates within hosts, and how it evades immune responses.
Overview: Virulence & Host Defense
We investigate how Toxoplasma gondii establishes chronic infection by identifying
parasite genes involved in virulence and host genes controlling susceptibility. Our lab integrates
CRISPR-based functional genomics, molecular parasitology, immunology, and live-cell imaging to dissect
host-pathogen interactions at high resolution.
Identifying Genes Required for In Vivo Fitness
To cause systemic infection, Toxoplasma must cross biological barriers, disseminate
efficiently, and evade innate immunity. It secretes virulence effectors, ROPs and
GRAs, from rhoptries and dense granules. Using genome-wide CRISPR loss-of-function
screens in vivo, we map parasite genes that mediate Toxoplasmosis pathogenesis.
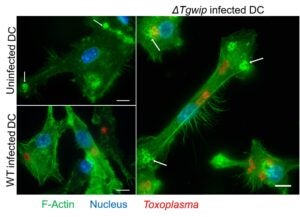
Dissemination Mechanisms
Toxoplasma dissemination is critical for neuropathology and chronic infection. In
collaboration with Antonio Barragan, we identified TgWIP, a secreted effector that
reprograms dendritic cell motility and facilitates dissemination. This work reveals how Toxoplasma
gondii hijacks host cell migration to promote disease progression.
Immune Evasion
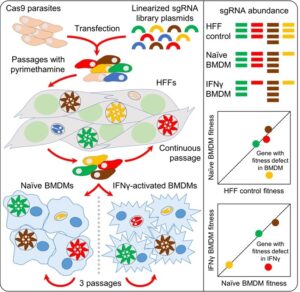
Macrophages are frontline defenders during infection. We performed a genome-wide
CRISPR screen that identified ~500 Toxoplasma genes affecting parasite fitness in
naïve and IFNγ-activated macrophages. A key hit, GRA45, prevents effector
aggregation during secretion. Parasites lacking GRA45 are hypersensitive to IFNγ and attenuated in
vivo.
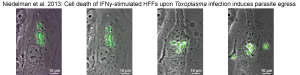
Proteins Driving Virulence in Human Cells
In human cells, immune mechanisms differ. ROP5/ROP18,
key in mice, are dispensable in humans. We identified new effectors required for
Toxoplasma fitness in IFNγ-stimulated human fibroblasts. For example, the parasite
effector TgIST prevents STAT1 dissociation from DNA, suppressing IFNγ-induced gene
expression.
Modulating Host Signaling
By infecting macrophages with 29 diverse strains and profiling transcriptomes, we revealed that
strain-specific effectors (e.g., ROP16, GRA15) differentially
activate STAT3/6 or NF-κB, modulating inflammation.
Host Genetics & Susceptibility
Using rat models, we found that resistant strains undergo rapid NLRP1-mediated macrophage death upon
infection, preventing parasite replication. Mouse macrophages activate inflammasomes without dying.
Frequently Asked Questions
What is Toxoplasmosis?
usually asymptomatic in healthy people but can be dangerous in pregnant women or immunocompromised
individuals.
How is Toxoplasmosis diagnosed?
IgG avidity testing helps determine timing of infection.
What are the treatment options?
These target acute infection, not latent cysts.
What does the Saeij Lab study?
pathogenesis, including immune evasion, dissemination, and host–pathogen interactions, using CRISPR
and comparative immunology.